Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
#1Re:Quantum Chemistry»A question about Molclus»2025-06-09 05:35:47
Dear Tian,
Thank you very much.
Sincerely,
Saeed
#2Re:Quantum Chemistry»A question about Molclus»2025-06-07 18:18:07
Dear Tian,
Your very kind attention and highly informative comments are deeply appreciated.
In the "genmer.ini" there is an option namely "ngen" associated with the number of configurations expected to be generated. it seems a high number is better since when number is increased, the chance of finding most stable conformed is also increased. For instanse, suppose I want to put 4 molecules of water around one molecule of HCl. Then, I select 10 for "ngen" indicating that 10 times or ten configurations should randomly be constructed any of which includes one HCl and four H2O. Again, I select 50 for "ngen". This time, 50 configurations will be constructed and undoubtedly the chance of an arrangement corresponding to the most stable conformer increases. Do you agree with me?
Best regards,
Saeed
#3Re:Quantum Chemistry»A question about Molclus»2025-06-06 19:49:53
Dear Tian,
Thank you very very much.
Please, if possible, let me ask another question:
When the "isostat" is executed, a question "Input the geometry threshold for distinguishing different clusters e.g. 0.25 (in Angstrom)" is asked. Please let me know what is the exact meaning of this question. I always enter 999 for this question. Is it correct?
Please, also let me know if "isuffle=1" in genmer.ini is always better than "ishuffle=0" and, leads to much better results.
Sincerely,
Saeed
#4Quantum Chemistry»A question about Molclus»2025-06-06 00:23:30
- saeed_E
- Replies: 6
-
Dear Tian,
The below command is given in the settings.ini of Molclus:
xtb_arg= "--gfn 1 --chrg 0 --uhf 0" // Additional arguments for xtb, e.g. "vtight --gfn 1 --gbsa h2o --chrg 0" (don't write task keywords such as "--opt" here)
Please suppose one wants to use xtb and employs the gfn2 method, while the structure is also optimized and the implicit solvent such as water is also considered. Please let me know how the above line should be changes to satisfy these expectations.
My own changes:
I changed the "igprog" to 4 and change itask to 3 so that xtb is used as computational code and optimization together with frequency is employed along any xtb execution. In addition, I changed the above line to the: xtb_arg= "--alpb water --gfn 2 --chrg 0 --uhf 0" // Additional arguments for xtb, e.g. "vtight --gfn 1 --gbsa h2o --chrg 0" (don't write task keywords such as "--opt" here).
Please let me know if all these changes are in line with my goal.
Best regards,
Saeed
#5Re:Quantum Chemistry»Molclus using for cluster optimization»2025-06-05 20:21:23
Dear Tian,
Thank you very much.
I will try to read your proposed examples and, if possible, I will again ask you provided that problems are encountered.
Sincerely,
Saeed
#6Quantum Chemistry»Molclus using for cluster optimization»2025-06-05 10:20:51
- saeed_E
- Replies: 2
-
Dear Tian,
Your very nice and powerful code, Molclus, can be used to find conformers resulting from dihedral angle rotations. Can this code be used to optimize a cluster of water molecules, including 5,6,... molecules? If so, could you please let me know and provide more explanation?
Best regards,
Saeed
#7Re:Multiwfn and wavefunction analysis»How to compute dipole moment for a spesific bond in a moleular entity»2025-06-01 07:24:53
Dear Tian,
Thank you very much.
Best,
Saeed
#8Multiwfn and wavefunction analysis»How to compute dipole moment for a spesific bond in a moleular entity»2025-05-26 16:46:33
- saeed_E
- Replies: 2
-
Dear Tian,
If possible, please let me know how to compute the value of the dipole moment for a given specific bond in a molecular entity using Multiwfn.
Best regards,
Saeed
#9Re:Quantum Chemistry»About sobEDA calculations....»2025-05-15 05:54:24
Dear Tian,
Many thanks for your very valuable guidance.
Best regards,
Saeed
#10Quantum Chemistry»About sobEDA calculations....»2025-05-14 15:27:13
- saeed_E
- Replies: 2
-
Dear Tian,
A moment ago, I calculated the value of interaction energy (delta_E_int) for an A...B complex optimized at B3LYP-D3(BJ)/6-311+G(2d,p) level using G16. However, sobEDA was also used to recalculate the interaction energy exactly at the same level. Since both levels are quite the same, I expected to obtain quite identical values for delta_E_int. But, there is a somewhat difference; that is, delta_E_int from G16 is -9.29 kcal/mol while delta_E_int from sobEDA is -8.82 kcal/mol, rendering a difference by 0.47 kcal/mol in absolute. Could you please let me know the source of such a difference?
Sincerely,
Saeed
#11Re:Multiwfn and wavefunction analysis»Performing ASM and sobEDA along whole IRC profile in a complete....»2025-05-14 15:18:43
Dear Tian,
OK. I am so happy and honored you liked my suggestion; thank you very much.
Please, also, let me indicate that I found a very inappreciable problem in one of the written codes. I am working on that to resolve this problem. Once this problem was resolved, I will try to prepare a detailed instruction (which is so time-consuming since I should explain one-by-one five scripts) and, if you let me, I will send them to you through your email.
Best regards,
Saeed
#12Multiwfn and wavefunction analysis»Performing ASM and sobEDA along whole IRC profile in a complete....»2025-05-13 00:28:09
- saeed_E
- Replies: 2
-
Dear Tian,
If possible, and you kindly let me, I would like to respectfully inform you that I can finally prepare nice bash scripts to perform activation/strain as well as sobEDA analyses along the whole IRC profile in a completely automated manner. If you find it appropriate, I can send these scripts with detailed instructions so that you (probably after some personalization and improving user experience), include them in the next releases of Multiwfn.
Best regards,
Saeed
#13Re:Quantum Chemistry»Using CDA to compute fragment orbitals responsible for Pauli»2025-04-28 09:11:07
Dear Tian,
Many thanks for your nice and professional comments.
Best regards,
Saeed
#14Re:Quantum Chemistry»Using CDA to compute fragment orbitals responsible for Pauli»2025-04-27 16:16:43
Dear Tian,
Your highly kind attention to prompt reply with much valuable and informative comments is extremely appreciated. Thank you very very much for the valuable time you so kindly assigned to my post.
Given your comments, do you have any other solutions to analyze Pauli repulsion between fragments in a given TS structure?
In addition, you have mentioned option 4 of CDA can directly give the overlap matrix. Isn't it necessary to first find the fragments' orbitals participating in the Pauli repulsion? Indeed, we should first find which fragment orbitals and in which complex orbital repeal each other due to Pauli repulsion.
Please also let me state that you cod "GauIRC2xyz" immediately crashes if the IRC.log file includes a TS comprising of three fragments (diene+dienophile+catalyst). Could you please change your code to be appropriate for three fragments?
Sincerely yours,
Saeed
#15Quantum Chemistry»Using CDA to compute fragment orbitals responsible for Pauli»2025-04-27 13:42:23
- saeed_E
- Replies: 4
-
Dear Tian,
As you nicely did guide me to use CDA for obtaining the value of Pauli repulsion between occupied orbitals of two interacting fragments (and, evaluating corresponding overlap), I did follow the below steps one-by-one. Please, if possible, let me know if my approach is quite correct:
1- The TS structure is fully optimized using, for instance, M062X/def2TZVPP level (TS includes two interacting fragments f1 and f2 which include some very heavy atoms such as Sn and, thus, I employed this basis set so that pseudo potentials are applied for such atoms).
2- After full optimization of TS, I changed number of atoms so that the electron-donor fragment (f1) to be the first fragment and electron-acceptor (f2) to be the second fragment.
3- Taking fully optimized TS structure, a SP is performed using "M062X/def2TZVPP nosymm pop=full iop(3/33=1)" keywords and, generated "chk" file is converted into "fch".
4- Taking the fully optimized structure of TS (whose atom numbering has been changed as described), f1 and f2 will be generated following a SP over each fragment using "M062X/def2-TZVPP nosymm pop=full" keywords and, "chk" files are converted into "fch" ones.
5-Multiwfn boots up, and the "fch" file of TS is loaded. Then 16 is chosen. I also enter 2 as there are two fragments. Then, the "fch" file of f1 (electron-donor) and "fch" file of f2 will be entered, respectively. The below data are presented:
Orb. Occ. d b d - b r
1 2.000000 -0.000003 0.000025 -0.000029 0.000014
2 2.000000 -0.000005 -0.000212 0.000206 -0.000071
3 2.000000 -0.000004 -0.000086 0.000082 -0.000026
4 2.000000 0.000445 -0.000000 0.000445 0.000024
5 2.000000 0.000304 -0.000001 0.000305 0.000043
6 2.000000 -0.000006 -0.000178 0.000171 -0.000065
7 2.000000 -0.000114 -0.001880 0.001766 0.022417
8 2.000000 0.005204 0.000709 0.004495 0.065189
9 2.000000 0.000370 0.002808 -0.002438 -0.003814
10 2.000000 -0.001802 -0.008178 0.006376 -0.011103
11 2.000000 0.003259 0.002135 0.001124 0.106547
12 2.000000 -0.005901 -0.000438 -0.005464 -0.101433
13 2.000000 -0.001358 -0.002066 0.000708 0.041290
14 2.000000 0.002171 -0.000691 0.002862 0.006158
15 2.000000 -0.001241 -0.001562 0.000321 -0.032240
16 2.000000 0.000064 0.002083 -0.002020 0.018551
17 2.000000 0.003422 -0.001551 0.004973 0.128120
18 2.000000 0.001397 -0.000052 0.001449 0.025455
19 2.000000 0.008062 -0.001752 0.009813 -0.089066
20 2.000000 0.018289 0.016942 0.001347 -0.159743
21 2.000000 0.051780 -0.002916 0.054697 -0.403971
22 2.000000 -0.000894 0.062648 -0.063542 -0.141167
-------------------------------------------------------------------
Sum: 44.000000 0.083437 0.065789 0.017649 -0.528892
As can be seen, the last four MO orbitals of complex (TS), 19-22, display a significant negative "r" value. So, one can conclude that the fragments orbitals participating in these MOs are responsible for the Pauli repulsion. Consequently, we should first decompose MOs 19-22 to the fragments orbitals. Now, we should select option "6, Decompose complex orbital contribution to CDA" to find which fragments orbital participate in the formation of MO 19. Once we select 6 followed by 19, and enter a threshold value such as 0.005, the following information is displayed:
Occupation number of orbital 19 of the complex: 2.00000000
FragA Orb(Occ.) FragB Orb(Occ.) d b d - b r
6( 2.0000) 7( 2.0000) 0.000000 0.000000 0.000000 -0.009978
6( 2.0000) 9( 2.0000) 0.000000 0.000000 0.000000 -0.026962
6( 2.0000) 10( 2.0000) 0.000000 0.000000 0.000000 -0.014043
6( 2.0000) 12( 2.0000) 0.000000 0.000000 0.000000 -0.021881
7( 2.0000) 7( 2.0000) 0.000000 0.000000 0.000000 -0.007323
7( 2.0000) 9( 2.0000) 0.000000 0.000000 0.000000 -0.005278
7( 2.0000) 12( 2.0000) 0.000000 0.000000 0.000000 0.007499
7( 2.0000) 13( 2.0000) 0.000000 0.000000 0.000000 -0.008777
Among NEGATIVE "r" values, the MOST NEGATIVE "r" is -0.026962 associated with a Pauli repulsion between orbital 6 of f1 and orbital 9 of f2. If we again repeat this calculation for orbitals 20, 21, 22 of TS and look for the most negative "r" value, we finally find that:
MO-19----> O-6_f1+O-9_f2
MO-20----> O-7_f1+O-13_f2
MO-21----> O-7_f1+O-13_f2
MO-22----> O-7_f1+O-15_f2
A simple visualization of the f1-orbitals (f1.fch is loaded into gview) one can see that orbitals 6 and 7 in f1 are HOMO-1 and HOM, respectively. These two orbitals are degenerate located over pi-orbitals of C-C triple bond of acetylene. On the other hand, a simple visualization of the f2-orbitals (f2.fch is loaded into gview), one can see that orbitals 9, 13, and 15 f2 are HOMO-6, HOMO-2, and HOMO, respectively. We should compute the value of S(i,j), the overlap integral, between:
orbital 6 of f1 and orbital 9 of f2
and
orbital 7 of f1 and each of orbitals 13 and 15 of f2.
To compute S(i,j) for f1 orbitals and f2 orbitals mentioned above, we should initially save TS.fch, f1.fch, and f2.fch as gaussian input files (gjf). Then, a SP is performed over TS.gjf using "M062X/def2TZVPP nosymm guess(save,only pop=none)" keywords and, corresponding chk file is converted into fch. A SP is also performed over each of f1.gjf and f2.gjf using "M062X/def2TZVPP nosymm" keywords and generated chk files are converted into fch ones. Now, Multiwfn boots up and TS.fch (newly generated as explained) is loaded. Then we select 100 followed by 15. The f1.fch and f2.fch is loaded, respectively and finally we enter 6,9. Then, we generate "whole overlap integral matrix to ovlpint.txt in current folder". From this file, one can see that:
S(6,9)= 0.056
S(7,13)= -0.126
S(7,15)= -0.031
We should consider the absolute value for negative S(I,j). Consequently, the Pauli repulsion between fragments orbitals at the TS structure increases as:
7,15<6,9<7,13
Please accept my highest apology for such a lengthy explanation.
Best Regards,
Saeed
#16Re:Quantum Chemistry»How to analyze Pauli repulsion with Multiwfn?»2025-04-27 04:14:26
Dear Tian,
Many many thanks for your highly valuable guidance.
Best regards,
Saeed
#17Quantum Chemistry»How to analyze Pauli repulsion with Multiwfn?»2025-04-26 16:55:01
- saeed_E
- Replies: 2
-
Dear Tian,
If possible and you kindly let me, I am going to repecfully ask a question.
Please, first, let me present a short explanation about what I am looking for. Consider a given Diels-Alder reaction which is further catalyzed with a catalyst. Suppose our analyses reveal that the catalyzed reaction is preferred over the uncatalyzed reaction due to the least Puli repulsion in the TS associated with the catalyzed reaction (I have performed an EDA such as sobEDA over both TSs to find this matter). Now, I want to find the appropriate occupied fragments orbitals at the TS structure, which are responsible for Pauli repulsion (for example HOMO-n to HOMO from one fragment vs. HOMO-m to HOMO of the other fragment). I do not know how Multiwfn able to perform such a nice analysis. In addition, I want to compute the overlap (S_i,j) between occupied orbitals of two fragments which are responsible for the most Pauli repulsion (If, for example, we can sort the pair orbitals responsibiling for the to 10 values of Pauli repulsion, S(I,j) needs to be computed in these top 10 interactions).
While I hope you kindly accept my highest apology for taking your valuable time with explanations, I would be highly grateful if you guide me to know how such a purpose can be reached by Multiwfn.
Best regards,
Saeed
#18Re:Quantum Chemistry»Sigme- or Pi-hole characterization with Laplacian of Rho»2025-03-23 03:14:31
Dear Tian,
Too many thanks for your kind attention and the highly valuable, informative, and professional suggestion.
Sincerely yours,
Saeed
#19Re:Quantum Chemistry»Sigme- or Pi-hole characterization with Laplacian of Rho»2025-03-22 19:52:10
Dear Tian,
Many thanks for your kind attention and, for guiding me with your highly valuable confirmation.
Please, also, let me ask one more question. In some cases, such as for TeHF compound in which the presence of a sigma-hole along the extension of the Te-F bond is expected, drawing isosurface of Laplacian of Rho (with isovalue= 0, namely the reactive surface) does not show a hole. On the other hand, this sigma-hole can be seen when isovalue is increased to 0.001 (a value slightly greater than zero). Could you please let me know why?
As another case, SnH3F could be mentioned. For this compound, the emergence of an expected hole along the Sn-F bond can never be satisfied using ANY value as isosurface for Laplacian of Rho. Such cases made me quite confused. Please let me know why some cases fail to present an expected sigma-hole over the Laplacian isosurface. I tried several times to attach "SnH3F.fch" file generated at M06-2X-D3(0)/def2-TZVPP level but, all tries failed! Using Multiwfn and VMD, I could not see the sigma-hole in this compound at any isovalue of Laplacian of Rho!
Sincerely,
Saeed
#20Quantum Chemistry»Sigme- or Pi-hole characterization with Laplacian of Rho»2025-03-21 18:39:05
- saeed_E
- Replies: 4
-
Dear Tian,
I hope you are doing well. Please, if possible, kindly let me ask you a question about Sigme- or Pi-hole characterization with the Laplacian of Rho.
Is it true to state that "a sigma-hole or a pi-hole is always characterized with the emergence of a hole on the isosurface of the Laplacian of Rho when an isovalue of zero is considered"?
In advance, thank you very much.
Sincerely,
Saeed
#21Re:Quantum Chemistry»An unexplainable observation in the EDA»2025-03-15 05:42:38
Dear Tian,
Many thanks for your highly valuable and informative comments.
Interestingly, as you have nicely recommended, the value of electrostatic interactions is quite consistent with the value of V_s,max calculated for heavy atoms in LAs.
Sincerely,
Saeed
#22Quantum Chemistry»An unexplainable observation in the EDA»2025-03-13 22:02:11
- saeed_E
- Replies: 2
-
Dear Tian,
Please, if possible, let me respectfully ask you a question regarding some unexplainable results in EDA I have recently encountered.
Please suppose 1,3-Aza Diene (A), including a sp2 hybridized nitrogen atom in position 2, interacts with some Lewis acids leading to inter-molecular tetrel-, pnictogen-, chalcogen-, and halogen-bound complexes (these LAs are SnH3F, SbH2F, TeHF, and FI all include a sigma-hole in the extension of heavy atom-F bond). These complexes participate in an Aza-Diles-Alder reaction toward a given dienophile (let's to be acetylene).
My energy decomposition energy analysis (EDA) indicates that electrostatic interaction is the main predominant player in stabilizing complexes considered. To explore the origin of the electrostatic interaction predominance, I calculated the atomic charge (Hirshfeld, NPA, VDD) on the heavy atoms of LAs. Unfortunately, computed atomic charges are not consistent with the values of electrostatic interactions. Indeed, while the positive charge on the heavy atoms of LAs decreases (becomes less positive) the electrostatic interaction between heavy atoms in LAs and nitrogen of Aza Diene increases (becomes more negative or stabilizing).
If possible, please let me know how you explain this strange and inconsistent observation.
In advance, please accept my highest gratitude for your kind attention to guiding me with your professional and golden comments and, please excuse me for bothering you.
Sincerely yours,
Saeed
#23Re:Multiwfn and wavefunction analysis»Electron Density Surfaces For Individual Localised Orbitals»2025-03-11 19:22:18
Thank you very much.
Saeed
#24Re:Multiwfn and wavefunction analysis»Electron Density Surfaces For Individual Localised Orbitals»2025-03-10 09:53:36
Dear Tian,
Many thanks for your much valuable comments.
If one wants to compare results of the gas phase and those of the solution, a quite same isovalue should be employed. In this sense, it seems isovalue= 0.001 a.u. to be suitable for both the gas and solution phase. Do you agree?
Sincerely,
Saeed
#25Re:Multiwfn and wavefunction analysis»Electron Density Surfaces For Individual Localised Orbitals»2025-03-09 20:22:18
Dear Tian,
In the above replies you indicated:
"the Bader's definition of molecular vdW surface (in gas phase) corresponds to isosurface of electron density with isovalue of 0.001 a.u.;.....". If possible, please let me know what is the suitable isovalue for the vdW electron density in the presence of a desirable solvent.
Sincerely yours,
Saeed
#26Re:Multiwfn and wavefunction analysis»Drawing BCPs and Laplacian isosurface both at the same time?»2025-03-05 11:42:37
Thank you very very much.
Best regards,
Saeed
#27Re:Quantum Chemistry»Does M05-2X also need correction like M06-2X?»2025-03-05 09:52:03
Dear Tian,
Too many thanks for your highly valuable and determining agreement and confirmation. In light of your highly valuable opinion, now I am ensured that my approach does not include any problem.
Sincerely,
Saeed
#28Re:Multiwfn and wavefunction analysis»Drawing BCPs and Laplacian isosurface both at the same time?»2025-03-05 09:46:48
Dear Tian,
Please accept my highest and deepest gratitude for the highly valuable time, energy, and patience you kindly spent to guide me in the best possible manner.
Following your instruction I could draw what I was looking for.
If possible, please let me ask a conceptual question. Considering my system I used "cub Laplacian 0.0" command in the VMD main windows so that the zero value is adjusted for both positive and negative parts of the Laplacian isosurface. As you can see in the presented picture, the selected BCP is located outside the zero isovalue of Laplacian. Consequently, it means the Laplacian at this BCP should be positive. Moreover, if a given BCP is located inside the zero isosurface of Laplacian (BCP is surrounded by Laplacian isosurface whose isovalue is zero), it means that Laplacian at that point should be negative. Please, if possible, let me know if my interpretations about the sign of Laplacian are reasonable based on how a BCP is located inside or outside of isosurface.
Sincerely yours,
Saeed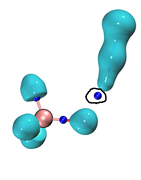
#29Quantum Chemistry»Does M05-2X also need correction like M06-2X?»2025-03-05 01:38:45
- saeed_E
- Replies: 2
-
Dear Tian,
It is well known that M06-2X functional has already been somewhat corrected during parametrization. So, to include dispersion corrections into this functional, one just needs to use "em=gd3". Please let me know has M05-2X also already been corrected to some extent during parametrization. If so, to include dispersion corrections, this functional also needs "em=gd3" keyword. Do you agree and confirm this statement?
In addition, I am computing solvation energy for a set of compounds at M05-2X/6-31G(d) level together with SMD solvation model as recommended by Truhlar and, as you also confirmed in one of your highly valuable blog articles. Please let me know if there are any problems regarding the accuracy of these calculations.
Sincerely yours,
Saeed
#30Re:Multiwfn and wavefunction analysis»Drawing BCPs and Laplacian isosurface both at the same time?»2025-03-04 09:41:59
Dear Tian,
Too many thanks for your so valuable guidance. But please let me state that I want to do visualization using a third party. Indeed, I want to generate corresponding "cub" files and, then, using "Cub" script in the VMD to reach very nice and high quality isosurface with BCPs. Could you please guide me to know how I can reach this purpose?
Sincerely,
Saeed