Multiwfn forum
Multiwfn official website: //www.umsyar.com/multiwfn. Multiwfn forum in Chinese: http://bbs.keinsci.com/wfn
You are not logged in.
- Topics:Active|Unanswered
Pages:1
#1Re:Quantum Chemistry»S1 optimization with negative frequency»2026-01-27 16:45:25
Dear Prof. Tian Lu,
Thank you for the information. I don't have access to Gaussian 16.
All the five imaginary frequencies belong to the rotation of individual methyl groups of t-butyl group or whole t-butyl group rotations only. Can I ignore them? If I run the ESD(ABS) for Full AH UV-vis spectrum or ESD(FLUOR), I need ESHessian. If ES Hessian derived from the geometry having multiple imaginary frequencies won't give error in my calculation?
Here is the output:
-----------------------
VIBRATIONAL FREQUENCIES
-----------------------
Scaling factor for frequencies = 1.000000000 (already applied!)
0: 0.00 cm**-1
1: 0.00 cm**-1
2: 0.00 cm**-1
3: 0.00 cm**-1
4: 0.00 cm**-1
5: 0.00 cm**-1
6: -144.10 cm**-1 ***imaginary mode***
7: -96.33 cm**-1 ***imaginary mode***
8: -71.26 cm**-1 ***imaginary mode***
9: -61.98 cm**-1 ***imaginary mode***
10: -40.99 cm**-1 ***imaginary mode***
11: 17.69 cm**-1
#2Quantum Chemistry»S1 optimization with negative frequency»2026-01-27 13:49:26
- Arivu
- Replies: 3
-
I am trying to optimize the S1 excited state geometry (input: GS geometry coordinates) with the following input.
"! LibXC(B3LYP) D3BJ RIJCOSX def2-SVP TightSCF TightOPT DEFGRID3 OPT FREQ
! PrintBasis MiniPrint PrintMOs
! AIM xyzfile pdbfile
%pal
nprocs 32
end
%maxcore 20000
%TDDFT
NROOTS 5
IROOT 1
FOLLOWIROOT TRUE
TDA FALSE
END
*xyz 0 1"
I ended up 5 imaginary frequencies. Am I doing any mistake. Any guidance in this problem is appreciated.
Thank you
#3Re:Quantum Chemistry»Native B3LYP vs LibXC(B3LYP) for S₀ and S₁ optimizations in ORCA (ESD»2026-01-21 07:43:07
Thank you so much for the clarifications Prof. Tian Lu.
#4Quantum Chemistry»Native B3LYP vs LibXC(B3LYP) for S₀ and S₁ optimizations in ORCA (ESD»2026-01-19 03:49:14
- Arivu
- Replies: 2
-
When I optimize the ground state (S₀) geometry in ORCA using the native B3LYP functional, the calculation runs without any problem.
However, when I attempt to optimize the first excited state (S₁), ORCA stops with an error and requests to use the LibXC implementation of B3LYP instead of the native one.
I would like to clarify the following points:
Are native B3LYP and LibXC(B3LYP) formally equivalent in ORCA, or are there meaningful differences in practice?
If LibXC(B3LYP) is required for excited-state optimizations (and for ESD calculations), is it recommended to also use LibXC(B3LYP) for the ground-state (S₀) optimization and frequency calculations to maintain methodological consistency?
Is it acceptable (from ORCA’s perspective and for publication-quality work) to optimize S₀ with native B3LYP and S₁ with LibXC(B3LYP), or should the same functional backend be used consistently for both states?
Any guidance on best practices for handling this situation would be greatly appreciated.
#5Re:Multiwfn and wavefunction analysis»Reorganization energy and Huang-Rhys factor»2026-01-09 17:41:03
Dear Prof. Tian Lu,
Thank you so much for the reply and nice suggestions.
#6Multiwfn and wavefunction analysis»Reorganization energy and Huang-Rhys factor»2026-01-03 16:52:14
- Arivu
- Replies: 2
-
Dear Prof. Tian Lu,
I am using Multiwfn for post-processing of DFT/TD-DFT results (mainly from ORCA) and would like to kindly clarify one point. Could you please confirm whether Multiwfn can be used to calculate reorganization energy and Huang–Rhys factors (including mode-resolved contributions) for electronic transitions (e.g., S₁ ↔ S₀)?
If not, is there any possibility of implementing this option in near future?
#7Re:Multiwfn and wavefunction analysis»Electron excitation analyses now exactly support TDDFT of ORCA!»2026-01-03 16:00:35
Dear Prof. Tian Lu,
Thank you so much. As you suggested it is working now after changing to ANSI.
#8Re:Multiwfn and wavefunction analysis»Electron excitation analyses now exactly support TDDFT of ORCA!»2025-12-27 14:45:12
Dear Prof. Tian Lu,
I have followed the exact procedure given in the website (//www.umsyar.com/758) of full TDDFT calculations. But I get a reply of the image attached when I choose the option of Enter as given below
"1 //Hole-electron analysis
[Enter] //Load an out file with the same name as the input file in the same directory (TDDFT.out)"
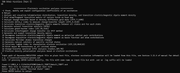
Kindly help to overcome this issue. I have all the necessary files but still problem.
Pages:1